
WEZENLA®
WEZENLA® (Ustekinumab) ist ein rein humaner monoklonaler IgG1κ-Antikörper gegen Interleukin (IL)-12/23 und wird für die Behandlung von Plaque-Psoriasis, Psoriatischer Arthritis und Morbus Crohn angewendet.
Kontakt
Fachinformationen
WEZENLA® (Ustekinumab) ist ein rein humaner monoklonaler IgG1κ-Antikörper gegen Interleukin (IL)-12/23 und wird für die Behandlung von Plaque-Psoriasis, Psoriatischer Arthritis und Morbus Crohn angewendet.
Noch kein DocCheck? Hier registrieren: www.doccheck.com
WEZENLA® (Ustekinumab) ist ein rein humaner monoklonaler IgG1κ-Antikörper gegen Interleukin (IL)-12/23.
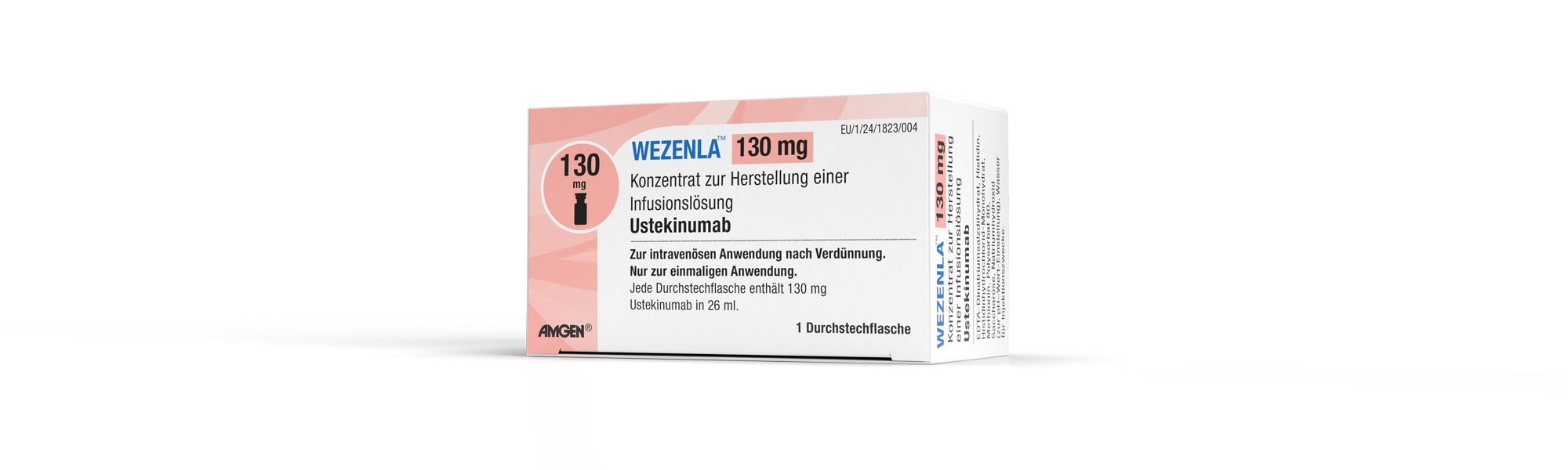
Die 130 mg Durchstechflasche wird für die Behandlung von Morbus Crohn angewendet.
Bezeichnung des Arzneimittels | Packungsgrößen | PZN |
| WEZENLA® 130 mg Konzentrat zur Herstellung einer Infusionslösung | 1 Durchstechflasche | 19279507 |
Amgen
WEZENLA® 130 mg Konzentrat zur Herstellung einer Infusionslösung
▼ Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden. Hinweise zur Meldung von Nebenwirkungen, siehe Abschnitt 4.8.
WEZENLA® 130 mg Konzentrat zur Herstellung einer Infusionslösung
Jede Durchstechflasche enthält 130 mg Ustekinumab in 26 ml (5 mg/ml).
Ustekinumab ist ein rein humaner monoklonaler IgG1κ-Antikörper gegen Interleukin (IL)-12/23, der unter Verwendung rekombinanter DNA-Technologie in einer Ovarialzelllinie des Chinesischen Hamsters produziert wird.
Sonstige Bestandteile mit bekannter Wirkung
Dieses Arzneimittel enthält 10,4 mg Polysorbat 80 (E 433) pro Dosiereinheit.
Vollständige Auflistung der sonstigen Bestandteile, siehe Abschnitt 6.1.
Konzentrat zur Herstellung einer Infusionslösung.
Die Lösung ist klar bis opaleszierend, farblos bis hellgelb. Die Lösung hat einen pH-Wert von etwa 6,0 und eine Osmolalität von etwa 280 mOsm/kg.
Morbus Crohn bei Erwachsenen
WEZENLA ist indiziert für die Behandlung erwachsener Patienten mit mittelschwerem bis schwerem aktiven Morbus Crohn, die entweder auf eine konventionelle Therapie oder einen der Tumornekrosefaktor-alpha (TNFα)-Antagonisten unzureichend angesprochen haben, nicht mehr darauf ansprechen oder eine Unverträglichkeit aufweisen.
Morbus Crohn bei Kindern und Jugendlichen
WEZENLA ist indiziert für die Behandlung von mittelschwerem bis schwerem aktiven Morbus Crohn bei Kindern und Jugendlichen mit einem Körpergewicht von mindestens 40 kg, die auf eine konventionelle Therapie oder auf ein Biologikum unzureichend angesprochen haben oder eine Unverträglichkeit aufweisen.
WEZENLA Konzentrat zur Herstellung einer Infusionslösung ist für die Anwendung unter der Anleitung und Überwachung eines in Diagnose und Behandlung des Morbus Crohn erfahrenen Arztes vorgesehen. WEZENLA Konzentrat zur Herstellung einer Infusionslösung darf nur für die intravenöse Induktionsdosis verwendet werden.
Dosierung
Erwachsene
Morbus Crohn
Die Behandlung mit WEZENLA ist mit einer auf dem Körpergewicht basierenden intravenösen Einzeldosis einzuleiten. Die Infusionslösung wird aus der in Tabelle 1 angegebenen Anzahl an Durchstechflaschen mit WEZENLA 130 mg zusammengestellt (zur Zubereitung siehe Abschnitt 6.6).
Tabelle 1. Initiale intravenöse Dosierung von WEZENLA
| Körpergewicht des Patienten zum Zeitpunkt der Dosierung | Empfohlene Dosisa | Anzahl der 130 mg WEZENLA- Durchstechflaschen |
| ≤ 55 kg | 260 mg | 2 |
| > 55 kg bis ≤ 85 kg | 390 mg | 3 |
| > 85 kg | 520 mg | 4 |
| Körpergewicht des Patienten zum Zeitpunkt der Dosierung | Empfohlene Dosisa | Anzahl der 130 mg WEZENLA- Durchstechflaschen |
| ≥ 40 kg bis ≤ 55 kg | 260 mg | 2 |
| > 55 kg bis ≤ 85 kg | 390 mg | 3 |
| > 85 kg | 520 mg | 4 |
Überempfindlichkeit gegen den Wirkstoff oder einen der in Abschnitt 6.1 genannten sonstigen Bestandteile.
Klinisch relevante, aktive Infektion (z. B. aktive Tuberkulose; siehe Abschnitt 4.4).
Rückverfolgbarkeit
Um die Rückverfolgbarkeit biologischer Arzneimittel zu verbessern, müssen die Bezeichnung des Arzneimittels und die Chargenbezeichnung des angewendeten Arzneimittels eindeutig dokumentiert werden.
Infektionen
Ustekinumab kann unter Umständen das Risiko von Infektionen erhöhen und latente Infektionen reaktivieren. In klinischen Studien und bei Psoriasis-Patienten in einer Beobachtungsstudie nach der Markteinführung wurden bei Patienten, die Ustekinumab erhielten, schwerwiegende bakterielle Infektionen, Pilz- und Virusinfektionen beobachtet (siehe Abschnitt 4.8).
Opportunistische Infektionen, darunter die Reaktivierung einer Tuberkulose, andere opportunistische bakterielle Infektionen (einschließlich atypischer Mykobakterieninfektion, Listerienmeningitis, Legionellenpneumonie und Nokardiose), opportunistische Pilzinfektionen, opportunistische Virusinfektionen (einschließlich durch Herpes simplex 2 verursachter Enzephalitis) und parasitäre Infektionen (einschließlich okulärer Toxoplasmose), wurden bei mit Ustekinumab behandelten Patienten gemeldet.
Bei Patienten mit einer chronischen Infektion oder einer rezidivierenden Infektion in der Vorgeschichte soll WEZENLA mit Vorsicht angewendet werden (siehe Abschnitt 4.3).
Vor Beginn der Behandlung mit WEZENLA sollen Patienten auf eine Tuberkuloseinfektion untersucht werden. WEZENLA darf Patienten mit aktiver Tuberkulose nicht verabreicht werden (siehe Abschnitt 4.3). Die Behandlung einer latenten Tuberkuloseinfektion muss vor Beginn der Behandlung mit WEZENLA eingeleitet werden. Eine Anti-Tuberkulosetherapie soll auch bei Patienten mit einer latenten oder aktiven Tuberkulose in der Vorgeschichte, bei denen ein angemessener Behandlungsverlauf nicht bestätigt werden kann, vor Behandlungsbeginn mit WEZENLA in Betracht gezogen werden. Patienten, die WEZENLA erhalten, müssen während und nach der Behandlung engmaschig auf Anzeichen und Symptome einer aktiven Tuberkulose überwacht werden.
Patienten sollen angewiesen werden, medizinischen Rat einzuholen, wenn Anzeichen oder Symptome auftreten, die auf eine Infektion hinweisen. Wenn ein Patient eine schwerwiegende Infektion entwickelt, muss der Patient engmaschig überwacht werden, und WEZENLA darf vor Abklingen der Infektion nicht verabreicht werden.
Maligne Tumoren
Immunsuppressiva wie Ustekinumab haben das Potenzial, das Risiko von malignen Tumoren zu erhöhen. Einige Patienten, die Ustekinumab in klinischen Studien erhielten, sowie Psoriasis-Patienten, die Ustekinumab in einer Beobachtungsstudie nach der Markteinführung erhielten, entwickelten kutane und nicht kutane maligne Tumoren (siehe Abschnitt 4.8). Das Risiko einer Malignität kann bei Psoriasis-Patienten, die im Verlauf ihrer Erkrankung mit anderen Biologika behandelt wurden, höher sein.
Es wurden keine Studien durchgeführt, in die Patienten mit malignen Tumoren in der Vorgeschichte eingeschlossen waren oder in denen die Behandlung bei Patienten fortgesetzt wurde, die einen malignen Tumor entwickelten, während sie Ustekinumab erhielten. Deshalb ist Vorsicht geboten, wenn eine Anwendung von Ustekinumab bei diesen Patienten in Erwägung gezogen wird.
Alle Patienten, besonders diejenigen über 60 Jahre sowie Patienten mit einer längeren immunsuppressiven Therapie oder PUVA-Behandlung in der Anamnese, sollten hinsichtlich des Auftretens von Hautkrebs überwacht werden (siehe Abschnitt 4.8).
Systemische und respiratorische Überempfindlichkeitsreaktionen
Systemisch
Nach Markteinführung wurde über schwerwiegende Überempfindlichkeitsreaktionen berichtet, in einigen Fällen mehrere Tage nach der Behandlung. Anaphylaxie und Angioödem traten auf. Wenn eine anaphylaktische oder eine andere schwerwiegende Überempfindlichkeitsreaktion auftritt, soll eine geeignete Therapie eingeleitet und die Verabreichung von WEZENLA abgebrochen werden (siehe Abschnitt 4.8).
Reaktionen im Zusammenhang mit einer Infusion
In klinischen Studien wurden Reaktionen im Zusammenhang mit einer Infusion beobachtet (siehe Abschnitt 4.8). Schwerwiegende Reaktionen im Zusammenhang mit einer Infusion, einschließlich anaphylaktischer Reaktionen auf die Infusion, wurden in der Zeit nach der Markteinführung gemeldet. Wenn eine schwerwiegende oder lebensbedrohliche Reaktion beobachtet wird, soll eine geeignete Therapie eingeleitet und Ustekinumab abgesetzt werden.
Respiratorisch
Nach Markteinführung wurden Fälle allergischer Alveolitis, eosinophiler Pneumonie und nicht-infektiöser organisierender Pneumonie während der Anwendung von Ustekinumab berichtet. Klinische Bilder umfassten Husten, Dyspnoe und interstitielle Infiltrate nach der Anwendung von einer bis drei Dosen. Zu den schwerwiegenden Folgen gehörten respiratorische Insuffizienz und Verlängerung des Krankenhausaufenthalts. Besserung wurde nach Absetzen von Ustekinumab und in einigen Fällen auch nach Verabreichung von Kortikosteroiden berichtet. Wenn eine Infektion ausgeschlossen und die Diagnose bestätigt wurde, sollte Ustekinumab abgesetzt und die entsprechende Behandlung durchgeführt werden (siehe Abschnitt 4.8).
Kardiovaskuläre Ereignisse
Kardiovaskuläre Ereignisse, einschließlich Myokardinfarkt und zerebrovaskulärer Insult, wurden bei Psoriasis-Patienten, die Ustekinumab erhielten, in einer Beobachtungsstudie nach der Markteinführung beobachtet. Die Risikofaktoren für kardiovaskuläre Erkrankungen sollten während der Behandlung mit WEZENLA regelmäßig überprüft werden.
Impfungen
Es wird nicht empfohlen, Lebendvirus- oder Lebendbakterienimpfstoffe (wie Bacillus Calmette Guérin (BCG)) gleichzeitig mit WEZENLA zu verabreichen. Mit Patienten, die kurz vorher Lebendvirus- oder Lebendbakterienimpfstoffe erhalten hatten, wurden keine spezifischen Studien durchgeführt. Zur sekundären Infektionsübertragung durch Lebendimpfstoffe bei Patienten, die Ustekinumab erhalten, liegen keine Daten vor. Vor einer Impfung mit Lebendviren oder lebenden Bakterien muss die Behandlung mit WEZENLA nach der letzten Dosis für mindestens 15 Wochen unterbrochen gewesen sein und kann frühestens 2 Wochen nach der Impfung wieder aufgenommen werden. Zur weiteren Information und Anleitung bezüglich der gleichzeitigen Anwendung von Immunsuppressiva nach der Impfung sollen die verordnenden Ärzte die Fachinformationen der spezifischen Impfstoffe hinzuziehen.
Die Verabreichung von Lebendimpfstoffen (z. B. der BCG-Impfstoff) an Säuglinge, die in utero gegenüber Ustekinumab exponiert waren, wird in den ersten zwölf Monaten nach der Geburt oder so lange nicht empfohlen, bis die Ustekinumab-Serumspiegel bei Säuglingen nicht nachweisbar sind (siehe Abschnitte 4.5 und 4.6). Wenn es einen eindeutigen klinischen Nutzen für den betroffenen Säugling gibt, kann die Verabreichung eines Lebendimpfstoffs zu einem früheren Zeitpunkt in Betracht gezogen werden, wenn die Ustekinumab-Serumspiegel beim Säugling nicht nachweisbar sind.
Patienten, die WEZENLA erhalten, können gleichzeitig Impfungen mit inaktivierten oder Totimpfstoffen erhalten.
Eine Langzeitbehandlung mit WEZENLA unterdrückt nicht die humorale Immunantwort auf Pneumokokken-Polysaccharid- oder Tetanusimpfstoffe (siehe Abschnitt 5.1).
Gleichzeitige Therapie mit Immunsuppressiva
In den Psoriasis-Studien wurden die Sicherheit und Wirksamkeit von Ustekinumab in Kombination mit Immunsuppressiva, einschließlich Biologika oder Phototherapie, nicht untersucht. In den Studien zur psoriatischen Arthritis schien die gleichzeitige Anwendung von MTX die Sicherheit oder Wirksamkeit von Ustekinumab nicht zu beeinflussen. In den Studien zu Morbus Crohn und Colitis ulcerosa schien die gleichzeitige Anwendung von Immunsuppressiva oder Kortikosteroiden die Sicherheit oder Wirksamkeit von Ustekinumab nicht zu beeinflussen. Wird die gleichzeitige Anwendung von anderen Immunsuppressiva und WEZENLA oder ein Wechsel von anderen biologischen Immunsuppressiva in Erwägung gezogen, ist Vorsicht geboten (siehe Abschnitt 4.5).
Immuntherapie
Ustekinumab wurde nicht bei Patienten untersucht, die sich einer Immuntherapie gegen eine Allergie unterzogen haben. Ob WEZENLA einen Einfluss auf eine Allergie-Immuntherapie hat, ist nicht bekannt.
Schwerwiegende Hautreaktionen
Bei Patienten mit Psoriasis wurde nach Behandlung mit Ustekinumab das Auftreten einer exfoliativen Dermatitis (Erythrodermie) berichtet (siehe Abschnitt 4.8). Bei Patienten mit Plaque-Psoriasis kann sich im Rahmen des natürlichen Verlaufs der Erkrankung eine erythrodermische Psoriasis entwickeln, deren Symptome sich klinisch möglicherweise nicht von denen einer exfoliativen Dermatitis unterscheiden. Im Rahmen der Psoriasis-Kontrolluntersuchungen müssen die Ärzte bei den Patienten auf Symptome einer erythrodermischen Psoriasis bzw. exfoliativen Dermatitis achten. Wenn entsprechende Symptome auftreten, muss eine angemessene Therapie eingeleitet werden. Bei Verdacht auf eine Arzneimittelreaktion muss WEZENLA abgesetzt werden.
Lupusbedingte Erkrankungen
Bei mit Ustekinumab behandelten Patienten wurden Fälle lupusbedingter Erkrankungen gemeldet, darunter kutaner Lupus erythematodes und Lupus-ähnliches Syndrom. Wenn Läsionen auftreten, insbesondere an sonnenexponierten Hautstellen oder zusammen mit einer Arthralgie, soll der Patient umgehend einen Arzt aufsuchen. Wenn die Diagnose einer lupusbedingten Erkrankung bestätigt wird, soll Ustekinumab abgesetzt und eine geeignete Behandlung eingeleitet werden.
Besondere Patientengruppen
Ältere Patienten (≥ 65 Jahre)
Bei Patienten ab 65 Jahren, die Ustekinumab erhielten, wurden im Vergleich zu jüngeren Patienten in klinischen Studien in den zugelassenen Indikationen keine Unterschiede in Bezug auf Sicherheit oder Wirksamkeit beobachtet. Die Anzahl der Patienten ab 65 Jahren ist jedoch nicht ausreichend, um feststellen zu können, ob sie im Vergleich zu jüngeren Patienten anders reagieren. Da es in der älteren Bevölkerung generell eine höhere Inzidenz von Infektionen gibt, ist bei der Behandlung von älteren Patienten Vorsicht geboten.
Natriumgehalt
WEZENLA enthält weniger als 1 mmol Natrium (23 mg) pro Durchstechflasche, d.h. es ist nahezu „natriumfrei“. Für die Infusion wird WEZENLA aber mit 9 mg/ml (0,9 %) Natriumchloridlösung verdünnt. Dies ist bei Patienten unter natriumkontrollierter Diät zu berücksichtigen (siehe Abschnitt 6.6).
Polysorbat 80
WEZENLA enthält 10,4 mg Polysorbat 80 (E 433) pro Dosiereinheit entsprechend 0,40 mg/ml. Polysorbate können allergische Reaktionen hervorrufen.
Lebendimpfstoffe sollen nicht zusammen mit WEZENLA gegeben werden.
Die Verabreichung von Lebendimpfstoffen (z. B. der BCG-Impfstoff) an Säuglinge, die in utero gegenüber Ustekinumab exponiert waren, wird in den ersten zwölf Monaten nach der Geburt oder so lange nicht empfohlen, bis die Ustekinumab-Serumspiegel bei Säuglingen nicht nachweisbar sind (siehe Abschnitte 4.4 und 4.6). Wenn es einen eindeutigen klinischen Nutzen für den betroffenen Säugling gibt, kann die Verabreichung eines Lebendimpfstoffs zu einem früheren Zeitpunkt in Betracht gezogen werden, wenn die Ustekinumab-Serumspiegel beim Säugling nicht nachweisbar sind.
In den populationspharmakokinetischen Analysen der Phase-3-Studien wurden die Auswirkungen der am häufigsten gleichzeitig bei Patienten mit Psoriasis angewendeten Arzneimittel (einschließlich Paracetamol, Ibuprofen, Acetylsalicylsäure, Metformin, Atorvastatin, Levothyroxin) auf die Pharmakokinetik von Ustekinumab untersucht. Es gab keine Hinweise auf eine Wechselwirkung mit diesen gleichzeitig verabreichten Arzneimitteln. Grundlage dieser Analyse war, dass mindestens 100 Patienten (> 5 % der untersuchten Population) über mindestens 90 % der Studiendauer gleichzeitig mit diesen Arzneimitteln behandelt wurden. Die Pharmakokinetik von Ustekinumab wurde nicht beeinflusst durch die gleichzeitige Anwendung von MTX, nichtsteroidalen Antirheumatika (NSARs), 6-Mercaptopurin, Azathioprin und oralen Kortikosteroiden bei Patienten mit psoriatischer Arthritis, Morbus Crohn oder Colitis ulcerosa. Weiterhin wurde die Pharmakokinetik von Ustekinumab nicht beeinflusst durch eine vorherige TNFα-Antagonisten-Exposition bei Patienten mit psoriatischer Arthritis oder Morbus Crohn oder eine vorherige Biologika-Exposition (z. B. TNFα-Antagonisten und/oder Vedolizumab) bei Patienten mit Colitis ulcerosa.
Die Ergebnisse einer In-vitro-Studie und einer Phase-1-Studie bei Patienten mit aktivem Morbus Crohn deuten nicht darauf hin, dass bei Patienten, die gleichzeitig CYP450-Substrate erhalten, eine Dosisanpassung erforderlich ist (siehe Abschnitt 5.2).
In den Psoriasis-Studien wurden die Sicherheit und Wirksamkeit von Ustekinumab in Kombination mit Immunsuppressiva, einschließlich Biologika oder Phototherapie, nicht untersucht. In den Studien zur psoriatischen Arthritis schien die gleichzeitige Anwendung von MTX die Sicherheit oder Wirksamkeit von Ustekinumab nicht zu beeinflussen. In den Studien zu Morbus Crohn und Colitis ulcerosa schien die gleichzeitige Anwendung von Immunsuppressiva oder Kortikosteroiden die Sicherheit oder Wirksamkeit von Ustekinumab nicht zu beeinflussen (siehe Abschnitt 4.4).
Frauen im gebärfähigen Alter
Frauen im gebärfähigen Alter müssen während und für mindestens 15 Wochen nach der Behandlung eine zuverlässige Verhütungsmethode anwenden.
Schwangerschaft
Daten aus einer moderaten Anzahl prospektiv erfasster Schwangerschaften nach Ustekinumab-Exposition mit bekanntem Ausgang, darunter mehr als 450 Schwangerschaften, bei denen die Exposition während des ersten Trimesters erfolgte, deuten nicht auf ein erhöhtes Risiko schwerer kongenitaler Fehlbildungen beim Neugeborenen hin.
Tierexperimentelle Studien ergaben keine Hinweise auf direkte oder indirekte gesundheitsschädliche Wirkungen in Bezug auf Schwangerschaft, embryonale/fetale Entwicklung, Geburt oder postnatale Entwicklung (siehe Abschnitt 5.3).
Die verfügbaren klinischen Erfahrungen sind jedoch begrenzt. Aus Vorsichtsgründen ist die Anwendung von WEZENLA während der Schwangerschaft möglichst zu vermeiden.
Ustekinumab ist plazentagängig und wurde im Serum von Säuglingen nach der Entbindung von Patientinnen nachgewiesen, die während der Schwangerschaft mit Ustekinumab behandelt wurden. Die klinischen Auswirkungen sind nicht bekannt, jedoch kann das Infektionsrisiko bei Säuglingen, die in utero gegenüber Ustekinumab exponiert waren, nach der Geburt erhöht sein. Die Verabreichung von Lebendimpfstoffen (z. B. der BCG-Impfstoff) an Säuglinge, die in utero gegenüber Ustekinumab exponiert waren, wird in den ersten zwölf Monaten nach der Geburt oder so lange nicht empfohlen, bis die Ustekinumab-Serumspiegel bei Säuglingen nicht nachweisbar sind (siehe Abschnitte 4.4 und 4.5). Wenn es einen eindeutigen klinischen Nutzen für den betroffenen Säugling gibt, kann die Verabreichung eines Lebendimpfstoffs zu einem früheren Zeitpunkt in Betracht gezogen werden, wenn die Ustekinumab-Serumspiegel beim Säugling nicht nachweisbar sind.
Stillzeit
Begrenzte Daten aus der veröffentlichten Literatur deuten darauf hin, dass Ustekinumab beim Menschen in sehr geringen Mengen in die Muttermilch übergeht. Es ist nicht bekannt, ob Ustekinumab nach der Aufnahme systemisch resorbiert wird. Aufgrund der Möglichkeit von unerwünschten Reaktionen bei gestillten Kindern muss eine Entscheidung darüber getroffen werden, ob das Stillen während und bis zu 15 Wochen nach der Behandlung zu unterbrechen ist oder ob die Behandlung mit WEZENLA zu unterbrechen ist. Dabei ist sowohl der Nutzen des Stillens für das Kind als auch der Nutzen der WEZENLA-Therapie für die Frau zu berücksichtigen.
Fertilität
Die Auswirkungen von Ustekinumab auf die Fertilität beim Menschen wurden nicht untersucht (siehe Abschnitt 5.3).
WEZENLA hat keinen oder einen zu vernachlässigenden Einfluss auf die Verkehrstüchtigkeit und die Fähigkeit zum Bedienen von Maschinen.
Zusammenfassung des Sicherheitsprofils
Die häufigsten Nebenwirkungen (> 5 %) in den kontrollierten Phasen der klinischen Studien mit Ustekinumab zu Psoriasis, psoriatischer Arthritis, Morbus Crohn und Colitis ulcerosa bei Erwachsenen waren Nasopharyngitis und Kopfschmerzen. Die meisten wurden als leicht eingestuft und erforderten keinen Abbruch der Studienmedikation. Die schwerwiegendsten Nebenwirkungen, die unter Ustekinumab berichtet wurden, waren schwerwiegende Überempfindlichkeitsreaktionen einschließlich Anaphylaxie (siehe Abschnitt 4.4). Das Gesamtsicherheitsprofil war bei Patienten mit Psoriasis, psoriatischer Arthritis, Morbus Crohn und Colitis ulcerosa ähnlich.
Tabellarische Auflistung der Nebenwirkungen
Die im Folgenden beschriebenen Daten zur Sicherheit geben die Ustekinumab-Exposition bei Erwachsenen in 14 Phase-2- und Phase-3-Studien mit 6 710 Patienten (4 135 mit Psoriasis und/oder psoriatischer Arthritis, 1 749 mit Morbus Crohn und 826 mit Colitis ulcerosa) wieder. Diese umfassen Ustekinumab-Expositionen in den kontrollierten und nicht-kontrollierten Phasen der klinischen Studien bei Patienten mit Psoriasis, psoriatischer Arthritis, Morbus Crohn oder Colitis ulcerosa über mindestens 6 Monate (4 577 Patienten) oder mindestens 1 Jahr (3 648 Patienten). 2 194 Patienten mit Psoriasis, Morbus Crohn oder Colitis ulcerosa wurden mindestens 4 Jahre behandelt, während 1 148 Patienten mit Psoriasis oder Morbus Crohn mindestens 5 Jahre behandelt wurden.
Tabelle 3 listet Nebenwirkungen aus klinischen Studien zu Psoriasis, psoriatischer Arthritis, Morbus Crohn und Colitis ulcerosa bei Erwachsenen sowie Nebenwirkungen, die nach Markteinführung berichtet wurden, auf. Die Nebenwirkungen sind nach Systemorganklasse und Häufigkeit unter Anwendung der folgenden Kategorien klassifiziert: sehr häufig (≥ 1/10), häufig (≥ 1/100, < 1/10), gelegentlich (≥ 1/1 000, < 1/100), selten (≥ 1/10 000, < 1/1 000), sehr selten (< 1/10 000), nicht bekannt (Häufigkeit auf Grundlage der verfügbaren Daten nicht abschätzbar). Innerhalb jeder Häufigkeitskategorie sind die Nebenwirkungen nach abnehmendem Schweregrad angegeben.
Tabelle 3. Liste der Nebenwirkungen
| Systemorganklasse | Häufigkeit: Nebenwirkung |
| Infektionen und parasitäre Erkrankungen | Häufig: Infektion der oberen Atemwege, Nasopharyngitis, Sinusitis Gelegentlich: Zellulitis, dentale Infektionen, Herpes zoster, Infektion der unteren Atemwege, Virusinfektion der oberen Atemwege, vulvovaginale Pilzinfektion |
| Erkrankungen des Immunsystems | Gelegentlich: Überempfindlichkeitsreaktionen (einschließlich Ausschlag, Urtikaria) Selten: schwerwiegende Überempfindlichkeitsreaktionen (einschließlich Anaphylaxie, Angioödem) |
| Psychiatrische Erkrankungen | Gelegentlich: Depression |
| Erkrankungen des Nervensystems | Häufig: Schwindelgefühl, Kopfschmerzen Gelegentlich: Fazialisparese |
| Erkrankungen der Atemwege, des Brustraums und Mediastinums | Häufig: oropharyngeale Schmerzen Gelegentlich: Nasenverstopfung Selten: allergische Alveolitis, eosinophile Pneumonie Sehr selten: organisierende Pneumonie* |
| Erkrankungen des Gastrointestinaltrakts | Häufig: Diarrhö, Übelkeit, Erbrechen |
| Erkrankungen der Haut und des Unterhautgewebes | Häufig: Pruritus Gelegentlich: pustulöse Psoriasis, Exfoliation der Haut, Akne Selten: exfoliative Dermatitis (Erythrodermie), Hypersensitivitätsvaskulitis Sehr selten: bullöses Pemphigoid, kutaner Lupus erythematodes |
| Skelettmuskulatur-, Bindegewebs- und Knochenerkrankungen | Häufig: Rückenschmerzen, Myalgie, Arthralgie Sehr selten: Lupus-ähnliches Syndrom |
| Allgemeine Erkrankungen und Beschwerden am Verabreichungsort | Häufig: Ermüdung/Fatigue, Erythem an der Injektionsstelle, Schmerzen an der Injektionsstelle Gelegentlich: Reaktionen an der Injektionsstelle (einschließlich Hämorrhagie, Hämatom, Verhärtung, Schwellung und Pruritus), Asthenie |
In klinischen Studien wurden Einzeldosen von bis zu 6 mg/kg intravenös ohne dosislimitierende Toxizität verabreicht. Im Falle einer Überdosierung wird empfohlen, den Patienten auf jegliche Anzeichen oder Symptome von Nebenwirkungen zu überwachen und gegebenenfalls umgehend eine geeignete symptomatische Behandlung einzuleiten.
Pharmakotherapeutische Gruppe: Immunsuppressiva, Interleukin-Inhibitoren. ATC-Code: L04AC05.
WEZENLA ist ein biologisch / biotechnologisch hergestelltes Arzneimittel, das im Wesentlichen einem bereits zugelassenen Arzneimittel gleicht. Ausführliche Informationen sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.
Wirkmechanismus
Ustekinumab ist ein rein humaner monoklonaler IgG1κ-Antikörper, der spezifisch an die gemeinsame p40-Protein-Untereinheit der humanen Zytokine Interleukin (IL)-12 und IL-23 bindet. Ustekinumab hemmt die Bioaktivität von humanem IL-12 und IL-23, indem es p40 daran hindert, an das IL-12Rß1-Rezeptorprotein, das auf der Oberfläche von Immunzellen exprimiert wird, zu binden. Ustekinumab kann nicht an IL-12 oder IL-23 binden, das bereits an IL-12Rß1-Zelloberflächenrezeptoren gebunden ist. Daher trägt Ustekinumab wahrscheinlich nicht zur Komplement- oder Antikörper-vermittelten Zytotoxizität der Zellen mit IL-12- und/oder IL-23-Rezeptoren bei. IL-12 und IL-23 sind heterodimere Zytokine, die von aktivierten Antigen-präsentierenden Zellen, wie Makrophagen und dendritischen Zellen, sezerniert werden. Beide Zytokine wirken an Immunfunktionen mit: IL-12 stimuliert natürliche Killerzellen (NK) und vermittelt die Differenzierung von CD4+ T-Zellen zum Phänotyp T-Helferzelle 1 (Th1), IL-23 induziert den T-Helfer-17(Th17)-Pfad. Eine anomale IL-12- und IL-23-Regulierung wurde mit immunvermittelten Krankheiten wie Psoriasis, psoriatischer Arthritis und Morbus Crohn assoziiert.
Es wird angenommen, dass Ustekinumab durch Bindung an die gemeinsame p40-Untereinheit von IL-12 und IL-23 seine klinischen Wirkungen bei Psoriasis, psoriatischer Arthritis und Morbus Crohn durch Unterbrechung der Th1- und Th17-Zytokinpfade entfaltet, die beide eine zentrale Rolle in der Pathologie dieser Krankheiten spielen.
Bei Patienten mit Morbus Crohn führte die Behandlung mit Ustekinumab während der Induktionsphase zu einer Abnahme von Entzündungsmarkern einschließlich C-reaktiven Proteins (CRP) und fäkalen Calprotectins, die während der gesamten Erhaltungsphase aufrechterhalten wurde. CRP wurde während der Studienverlängerung bestimmt, und die während der Erhaltungsphase beobachteten Reduktionen blieben im Allgemeinen bis Woche 252 erhalten.
Immunisierung
Während der Verlängerungsphase der Psoriasis-Studie 2 (PHOENIX 2) zeigten die über mindestens 3,5 Jahre mit Ustekinumab behandelten erwachsenen Patienten eine ähnliche Antikörperantwort auf Pneumokokken-Polysaccharid und Tetanus-Impfstoffe wie die nicht systemisch behandelten Psoriasispatienten in der Kontrollgruppe. Bei den mit Ustekinumab behandelten erwachsenen Patienten und der Kontrollgruppe war der Anteil der Patienten, der protektive Antipneumokokken- und Antitetanus-Antikörper entwickelte, vergleichbar. Auch die Antikörpertiter waren bei den mit Ustekinumab behandelten Patienten und der Kontrollgruppe vergleichbar.
Klinische Wirksamkeit und Sicherheit
Morbus Crohn
Die Sicherheit und Wirksamkeit von Ustekinumab wurden in drei randomisierten, doppelblinden, placebokontrollierten multizentrischen Studien mit erwachsenen Patienten mit mittelschwerem bis schwerem aktiven Morbus Crohn (Crohn’s Disease Activity Index[CDAI]-Score von ≥ 220 und ≤ 450) untersucht. Das klinische Entwicklungsprogramm bestand aus zwei 8-wöchigen Studien zur intravenösen Induktion (UNITI-1 und UNITI-2), gefolgt von einer 44-wöchigen randomisierten Studie zur subkutanen Erhaltungstherapie (IM-UNITI; randomized withdrawal maintenance study), was insgesamt einer Therapiedauer von 52 Wochen entspricht.
In die Induktionsstudien waren 1 409 Patienten eingeschlossen (UNITI-1, n = 769; UNITI-2, n = 640). Der primäre Endpunkt in beiden Induktionsstudien war der Anteil der Patienten mit klinischem Ansprechen (definiert als Abnahme des CDAI-Scores um ≥ 100 Punkte) in Woche 6. Daten zur Wirksamkeit wurden von beiden Studien bis einschließlich Woche 8 erfasst und ausgewertet. Gleichzeitige Gaben von oralen Kortikosteroiden, Immunmodulatoren, Aminosalicylaten und Antibiotika waren erlaubt, und 75 % der Patienten erhielten weiterhin mindestens eine dieser Medikationen. In beiden Studien erhielten die Patienten in Woche 0 randomisiert eine einmalige intravenöse Gabe entweder entsprechend der empfohlenen auf dem Körpergewicht basierenden Dosis von etwa 6 mg/kg (siehe Tabelle 1, Abschnitt 4.2), eine Fixdosis von 130 mg Ustekinumab oder Placebo.
Die Patienten in UNITI-1 hatten auf eine oder mehrere vorherige Anti-TNFα-Therapien nicht angesprochen oder diese nicht vertragen. Etwa 48 % der Patienten hatten auf 1 vorherige Anti-TNFα-Therapie und 52 % auf 2 oder 3 vorherige Anti-TNFα-Therapien nicht angesprochen. 29,1 % der Patienten dieser Studie hatten dabei initial unzureichend angesprochen (primäre Non-Responder); 69,4 % hatten ein Ansprechen, das jedoch verloren ging (sekundäre Non-Responder), und 36,4 % hatten die Anti-TNFα-Therapien nicht vertragen.
Die Patienten in UNITI-2 hatten auf mindestens eine konventionelle Therapie, einschließlich Kortikosteroiden und Immunmodulatoren, nicht angesprochen und waren entweder Anti-TNFα-naiv (68,6 %) oder hatten vorher eine Anti-TNFα-Therapie erhalten und auf diese auch angesprochen (31,4 %).
Sowohl in UNITI-1 als auch UNITI-2 war der Anteil der Patienten mit klinischem Ansprechen und Remission in der mit Ustekinumab behandelten Gruppe im Vergleich zu Placebo signifikant größer (siehe Tabelle 4). Klinisches Ansprechen und Remission waren in den mit Ustekinumab behandelten Patienten bereits in Woche 3 signifikant und nahmen bis einschließlich Woche 8 weiter zu. In diesen Induktionsstudien war die Wirksamkeit in der Gruppe mit der auf dem Körpergewicht basierenden Dosis größer und länger anhaltend als in der Gruppe mit der 130-mg-Dosis. Deshalb ist die auf dem Körpergewicht basierende Dosis die empfohlene Dosis für die intravenöse Induktion.
Tabelle 4. Induktion von klinischem Ansprechen und Remission in UNITI-1 und UNITI-2
| UNITI-1* | UNITI-2** | |||
| Placebo n = 247 | Empfohlene Ustekinumab- dosis n = 249 | Placebo n = 209 | Empfohlene Ustekinumab- dosis n = 209 | |
| Klinische Remission, Woche 8 | 18 (7,3 %) | 52 (20,9 %)a | 41 (19,6 %) | 84 (40,2 %)a |
| Klinisches Ansprechen (100 Punkte), Woche 6 | 53 (21,5 %) | 84 (33,7 %)b | 60 (28,7 %) | 116 (55,5 %)a |
| Klinisches Ansprechen (100 Punkte), Woche 8 | 50 (20,2 %) | 94 (37,8 %)a | 67 (32,1 %) | 121 (57,9 %)a |
| 70-Punkte-Ansprechen, Woche 3 | 67 (27,1 %) | 101 (40,6 %)b | 66 (31,6 %) | 106 (50,7 %)a |
| 70-Punkte-Ansprechen, Woche 6 | 75 (30,4 %) | 109 (43,8 %)b | 81 (38,8 %) | 135 (64,6 %)a |
| Placebo* n = 131† | 90 mg Ustekinumab alle 8 Wochen n = 128† | 90 mg Ustekinumab alle 12 Wochen n = 129† | |
| Klinische Remission | 36 % | 53 %a | 49 %b |
| Klinisches Ansprechen | 44 % | 59 %b | 58 %b |
| Kortikosteroidfreie klinische Remission | 30 % | 47 %a | 43 %c |
| Klinische Remission bei Patienten, | |||
| die zu Beginn der Erhaltungstherapie in Remission waren | 46 % (36/79) | 67 % (52/78)a | 56 % (44/78) |
| aus Studie CRD3002‡ | 44 % (31/70) | 63 % (45/72)c | 57 % (41/72) |
| die Anti-TNFα-naiv sind | 49 % (25/51) | 65 % (34/52)c | 57 % (30/53) |
| aus Studie CRD3001§ | 26 % (16/61) | 41 % (23/56) | 39 % (22/57) |
| 90 mg Ustenikumab alle 8 Wochen N = 23 | 90 mg Ustekinumab alle 12 Wochen N = 25 | Gesamtzahl der Patienten N = 48 | |
| Klinische Remission* | 43,5 % (10/23) | 60,0 % (15/25) | 52,1 % (25/48) |
| Kortikosteroidfreie klinische Remission§ | 43,5 % (10/23) | 60,0 % (15/25) | 52,1 % (25/48) |
| Klinische Remission bei Patienten, die in Induktionswoche 8 in klinischer Remission waren* | 64,3 % (9/14) | 54,5 % (6/11) | 60,0 % (15/25) |
| Klinisches Ansprechen† | 52,2 % (12/23) | 60,0 % (15/25) | 56,3 % (27/48) |
| Endoskopisches Ansprechen£ | 22,7 % (5/22) | 28,0 % (7/25) | 25,5 % (12/47) |
Nach der empfohlenen intravenösen Induktionsdosis betrug die mediane Spitzenkonzentration von Ustekinumab im Serum, beobachtet eine Stunde nach Infusion, 126,1 μg/ml bei Patienten mit Morbus Crohn.
Verteilung
Das mediane Verteilungsvolumen lag in der terminalen Phase (Vz) nach einer einzelnen intravenösen Gabe bei Patienten mit Psoriasis zwischen 57 und 83 ml/kg.
Biotransformation
Der genaue Stoffwechselweg von Ustekinumab ist nicht bekannt.
Elimination
Die mediane systemische Clearance (CL) lag nach einer einzelnen intravenösen Verabreichung an Patienten mit Psoriasis zwischen 1,99 und 2,34 ml/Tag/kg. Die mediane Halbwertszeit (t1/2) von Ustekinumab betrug bei Patienten mit Morbus Crohn, Psoriasis und/oder psoriatischer Arthritis ungefähr 3 Wochen und bewegte sich in allen Studien zu Psoriasis und psoriatischer Arthritis zwischen 15 und 32 Tagen.
Dosislinearität
Die systemische Verfügbarkeit von Ustekinumab (Cmax und AUC) erhöhte sich ungefähr dosisproportional nach einer einzelnen intravenösen Verabreichung von Dosen zwischen 0,09 mg/kg und 4,5 mg/kg.
Besondere Patientengruppen
Es liegen keine pharmakokinetischen Daten von Patienten mit Nieren- oder Leberfunktionsstörung vor.
Es wurden keine spezifischen Studien mit intravenösem Ustekinumab mit älteren Patienten oder Kindern und Jugendlichen mit einem Körpergewicht von unter 40 kg durchgeführt.
Bei Patienten mit Morbus Crohn wurde die Variabilität der Clearance von Ustekinumab durch Körpergewicht, Serumkonzentration von Albumin, Geschlecht und Antikörperstatus gegenüber Ustekinumab beeinflusst; dabei war das Körpergewicht die wichtigste Kovariable, die das Verteilungsvolumen beeinflusste. Zusätzlich wurde die Clearance bei Morbus Crohn durch C-reaktives Protein, den TNFα-Antagonisten-Versagerstatus und ethnische Zugehörigkeit (asiatisch versus nicht-asiatisch) beeinflusst. Der Einfluss dieser Kovariablen lag innerhalb von ±20 % des typischen oder Referenzwertes des jeweiligen PK-Parameters, sodass eine Dosisanpassung für diese Kovariablen nicht gerechtfertigt ist. Die gleichzeitige Anwendung von Immunmodulatoren hatte keinen signifikanten Effekt auf die Disposition von Ustekinumab.
Regulierung von CYP450-Enzymen
Die Auswirkungen von IL-12 oder IL-23 auf die Regulierung von CYP450-Enzymen wurden in einer In-vitro-Studie an humanen Hepatozyten untersucht. Sie zeigte, dass IL-12 und/oder IL-23 bei Konzentrationen von 10 ng/ml die humanen CYP450-Enzymaktivitäten (von CYP1A2, 2B6, 2C9, 2C19, 2D6 oder 3A4) nicht ändern (siehe Abschnitt 4.5).
In einer offenen Phase-1-Studie zu Arzneimittelwechselwirkungen (Studie CNTO1275CRD1003) wurde der Effekt von Ustekinumab auf die Aktivitäten von Cytochrom-P450-Enzymen nach der Induktions- und Erhaltungsdosis bei Patienten mit aktivem Morbus Crohn (n = 18) untersucht. Es wurden keine klinisch signifikanten Veränderungen in der Exposition gegenüber Koffein (CYP1A2-Substrat), Warfarin (CYP2C9-Substrat), Omeprazol (CYP2C19-Substrat), Dextromethorphan (CYP2D6-Substrat) oder Midazolam (CYP3A-Substrat) bei gleichzeitiger Anwendung von Ustekinumab in der zugelassenen empfohlenen Dosierung bei Patienten mit Morbus Crohn beobachtet (siehe Abschnitt 4.5).
Kinder und Jugendliche
Die Ustekinumab-Serumkonzentrationen bei Kindern und Jugendlichen mit Morbus Crohn und einem Körpergewicht von mindestens 40 kg, die mit der empfohlenen gewichtsbasierten Dosis behandelt wurden, waren im Allgemeinen mit der erwachsener Patienten mit Morbus Crohn vergleichbar, die mit der gewichtsbasierten Dosis für Erwachsene behandelt wurden.
Basierend auf den Studien zur Sicherheitspharmakologie, Toxizität bei wiederholter Gabe, Entwicklungs- und Reproduktionstoxizität lassen die präklinischen Daten keine besonderen Gefahren (z. B. Organtoxizität) für den Menschen erkennen. In Entwicklungs- und Reproduktionstoxizitätsstudien mit Cynomolgus-Affen wurden weder unerwünschte Wirkungen auf die männlichen Fertilitätsindices noch Geburtsdefekte oder Entwicklungstoxizität beobachtet. Bei Anwendung eines analogen IL-12/23-Antikörpers bei Mäusen wurden keine unerwünschten Wirkungen auf die weiblichen Fertilitätsindices beobachtet.
Die Dosen in tierexperimentellen Studien waren bis zu ca. 45-fach höher als die höchste äquivalente Dosis, die Psoriasis-Patienten verabreicht werden soll, und resultierten bei Affen in mehr als 100-fach höheren als die bei Menschen beobachteten Spitzenkonzentrationen im Serum.
Studien zur Karzinogenität wurden mit Ustekinumab aufgrund des Mangels an geeigneten Modellen für einen Antikörper ohne Kreuzreaktivität auf Nager-IL-12/23-p40 nicht durchgeführt.
EDTA-Dinatriumsalzdihydrat
Histidin
Histidinhydrochlorid-Monohydrat
Methionin
Polysorbat 80 (E 433)
Saccharose
Natriumhydroxid (zur pH-Wert-Einstellung)
Wasser für Injektionszwecke
Da keine Kompatibilitätsstudien durchgeführt wurden, darf dieses Arzneimittel nicht mit anderen Arzneimitteln gemischt werden. WEZENLA darf nur mit 9 mg/ml (0,9 %) Natriumchloridlösung verdünnt werden. WEZENLA soll nicht gleichzeitig mit anderen Arzneimitteln über dasselbe intravenöse Schlauchsystem gegeben werden.
Im Kühlschrank lagern (2 °C – 8 °C).
Nicht einfrieren.
Die Durchstechflasche im Umkarton aufbewahren, um den Inhalt vor Licht zu schützen.
Aufbewahrungsbedingungen nach Verdünnung des Arzneimittels, siehe Abschnitt 6.3.
26 ml Lösung in einer 30-ml-Durchstechflasche aus Typ-I-Glas, die mit einem Elastomerstopfen verschlossen ist.
WEZENLA ist in einer Packung mit 1 Durchstechflasche erhältlich.
Die Lösung in der WEZENLA-Durchstechflasche darf nicht geschüttelt werden. Die Lösung muss vor der Verabreichung visuell auf Schwebstoffe oder Verfärbung überprüft werden. Die Lösung ist klar bis opaleszierend, farblos bis hellgelb. Das Arzneimittel darf nicht verwendet werden, wenn die Lösung verfärbt oder trübe ist oder Schwebstoffe aufweist.
Verdünnung
WEZENLA Konzentrat zur Herstellung einer Infusionslösung muss von medizinischem Fachpersonal unter aseptischen Bedingungen verdünnt und zubereitet werden.
1. Berechnen Sie die Dosis und die Anzahl der benötigten WEZENLA-Durchstechflaschen auf Basis des Körpergewichts des Patienten (siehe Abschnitt 4.2, Tabelle 1). Jede 26-ml-Durchstechflasche WEZENLA enthält 130 mg Ustekinumab. Verwenden Sie immer das gesamte Volumen der WEZENLA-Durchstechflaschen.
2. Entnehmen Sie dem 250-ml-Infusionsbeutel ein Volumen der 9 mg/ml (0,9 %) Natriumchloridlösung, das dem hinzuzufügenden Volumen von WEZENLA entspricht und verwerfen Sie es. (Verwerfen Sie 26 ml Natriumchloridlösung für jede benötigte WEZENLA-Durchstechflasche. Bei 2 Durchstechflaschen verwerfen Sie 52 ml, bei 3 Durchstechflaschen 78 ml und bei 4 Durchstechflaschen 104 ml).
3. Ziehen Sie aus jeder benötigten Durchstechflasche 26 ml WEZENLA auf und fügen Sie diese dem 250-ml-Infusionsbeutel hinzu, das endgültige Volumen im Infusionsbeutel soll 250 ml betragen. Mischen Sie die Lösung behutsam.
4. Prüfen Sie die verdünnte Lösung vor der Gabe visuell. Verwenden Sie sie nicht, wenn sie opake Partikel, Verfärbungen oder Schwebstoffe aufweist.
5. Verabreichen Sie die Infusionslösung über einen Zeitraum von mindestens einer Stunde.
6. Verwenden Sie nur ein Infusionsset mit einem sterilen, nicht pyrogenen In-line-Filter mit geringer Proteinbindung (Porengröße 0,2 Mikrometer).
7. Jede Durchstechflasche ist nur für den einmaligen Gebrauch bestimmt. Nicht verwendetes Arzneimittel oder Abfallmaterial ist entsprechend den nationalen Anforderungen zu beseitigen.
Amgen Technology (Ireland) UC,
Pottery Road,
Dun Loaghaire,
Co Dublin,
Irland
EU/1/24/1823/004
Datum der Erteilung der Zulassung: 20. Juni 2024
Juli 2025
Ausführliche Informationen zu diesem Arzneimittel sind auf den Internetseiten der Europäischen Arzneimittel-Agentur https://www.ema.europa.eu verfügbar.
Verschreibungspflichtig
1 Durchstechflasche
Amgen GmbH
Riesstraße 24
80992 München
Tel.: 089 149096 0
Fax: 089 149096 2000
www.amgen.de
MedInfo-Hotline: 0800 - 264 36 44
medinfo.amgen.de